If you are thinking I am the person to send InsightII tutorials to, please see my tutorials page.
5/14/07:
Again in regard to the availability of grades, we have determined that the
second (Wednesday) section's grades are not up via the Registrar's website
(which I believe is what would do myRutgers grades/transcripts),
and neither are the grades for a number of other classes (with other
professors) - evidently their computer messed up somehow. We are working on
getting the grades in via a paper copy (the Registrar's computer won't
accept them electronically now) and will try to also get them up on the FAS
gradebook. Sorry about this; we had thought all the grades would be
available, but (since I doubt multiple professors did something wrong when
uploading them) it appears the Registrar messed up...
5/11/07:
In regard to the availability of grades, I am hearing that people are not
seeing their grades on the FAS gradebook, probably because Dr. Chase put the
grades into the Registrar's website (not into the FAS gradebook, since Cook
is not part of FAS), which appears not to be completely linked with the FAS
gradebook. At least some people have seen their grades somehow; I am
guessing this was done by using the registrar's website, but am not sure. It
is possible that they are expediting the grades for (hopefully!) graduating
seniors; however, whose grades are showing up appears to be rather
inconsistent. If the grades are not up Monday, we will make further
enquiries into what the problem is.
In regard to turnitin.com percentages, I have received a couple of queries as to why people are being accused of cheating when the percentage is not extremely high. The percentages are only used as an indication of what lab reports to initially look at for possible problems.
For those currently waiting around for Dr. Chase due to TFs due to evidence of cheating (there are 4 people with TFs for this reason, BTW), I suggest emailing Dr. Chase over the weekend (chase_c at aesop dot rutgers dot edu) for an appointment no earlier than Monday. He is currently working on grading Proteins and Enzymes final papers. Bugging him will decrease the chances of any leniency, not increase them.
For those who thought they were graduating and have received TFs due to evidence of cheating, there are two possibilities; I am not sure which is the case:
In regard to people with grades lower than they might have expected from a 90-100 == A system:
People should note that, even if your lab reports have been returned to you, this does not mean that the grade can't change. Reasons for it changing range from if you did something over again and had it regraded (only with permission, for a few of the plasmid labs for which people misunderstood completely what they were doing - get these back by the 15th of May, please...) to turnitin.com findings (which, if we decide they are significant - if you have a TF, then we have concluded that they are indeed significant; in other cases, the matter is as yet uncertain (see next paragraph) - will be turned over to the judicial system after Dr. Chase finishes working on grades for Proteins and Enzymes). In either of these cases, you may have a T grade; you may also have a T grade if your lab reports were turned in very late and have not been graded as yet, of course. (For people planning on graduating this month, yes, we will do our best to expedite any regrading, etcetera to take this into account - but, in the case of the judicial system, its speed is not entirely up to us...)
In regard to the judicial system and turnitin.com results, note that said results are not the only material examined. Also note that not everyone whose lab reports were handed back late will necessarily be charged under the judicial system, depending on both why the lab report was handed back late (e.g., if it wasn't handed in on time - either on paper or on turnitin.com - that would be a reason why it wouldn't be handed back with the others, even if the turnitin.com results for it were fine) and the level of evidence available. Said level of evidence, however, can change due to the testimony of both people charged and anyone uninvolved who the people charged can get to testify. (It would not be fair to penalize only some people involved; I therefore encourage submission of further evidence - provided that it can be backed up, either by testimony from someone not accused (and not otherwise biased) or from turnitin.com or, preferably, both - so that others involved are likewise penalized.)
5/5/07:
A new example sequencing question (note that the sequences differ for each
person; they are computer-generated, by a program I wrote): expbio.question.example2.txt. The
old example (expbio.question.example.txt) is still
valid as an example, but the phrasing and point totals are more up to date
in the expbio.question.example2.txt version.
Example plasmid assembly questions (note that each person's question will vary; again, they are computer-generated, by a program I wrote):
The exams from IRIX may be accessible via:
For other studying for the final, please see my comments.
The following people have plasmid and/or sequencing lab reports now available in my office (last names removed/shortened for privacy reasons); I will be here until around 6 PM (sorry, not 7):
I will next be working on the labs of those who have submitted
turnitin.com data late.
5/2/07:
I am, as stated before, trying to get all the plasmid and sequencing labs
graded by the time of the general biochem exam (unless they were in
extremely late or for which there is reasonable suspicion of
cheating (including being copied off of)), so that people can have them
to study by for the experimental exam. DO NOT DISTURB!. If
you need to turn in something, put it underneath my door or give it to Dr.
Chase. Email me if you need to ask questions; I may or may not answer today!
I have put upstairs in Dr. Chase's office the plasmid labs that I have
graded and have not yet seen people to give back (Alex L, Aniket R, Joanna
T, Blake S, Doug N, Jigi P, Saddef H, Shreya S, Yolanda P, Illysa B,
Geoffery W, Natallia K). The grades so far on the plasmid lab are... highly
variable.
I have graded the sequencing labs, with exceptions as noted above (plus some for which the people have not turned them in on turnitin.com); the grades are likewise highly variable (max 104, min 59, median 89, mean 86.64). Please, if you got anything significantly wrong in the questions for the sequencing labs, go over dideoxynucleotide sequencing before the final, which will have a sequencing question on it (worth at minimum 10 points - possibly 15 points...). I will give the graded sequencing labs to Dr. Kahn or Dr. VanEs to take to the general biochem final; ditto with the plasmid labs that people have not picked up yet.
4/30/07:
There is no turnitin.com entry for the RNA lab.
I am trying to get all of the plasmid and sequencing labs graded by the time of the general biochem exam (unless they were in extremely late), so that people can have them to study by for the experimental exam.
4/29/07:
I will be in today until 10 PM or so, but please avoid disturbing me
(further ;-}) except to hand labs in, if possible. I am not putting up a
full-scale "do not disturb" sign, but would much prefer helping people via
email (not in-person or via phone calls, especially not the latter!) as much
as possible.
With regard to the data analysis section in the RNA lab, the part about the "fraction" is confusing. For the cDNA, you used 1 ug of mRNA per 20 ul of solution. This works out to a concentration of 0.05 ug/ul. Multiply that by the 2 ul (approximately) that you used per well (unless you used 6.3 ul in less than 3 wells), and you get the amount of RNA used in the reaction (you then take the amount of the _particular_ mRNA that we detected, in moles, and divide it by that to get the ratio).
4/28/07:
I am needing to be alone to get some work done. Do not disturb.
4/27/07:
It is OK on the RNA lab to give the results in terms of
(femtomoles|picomoles|attomoles) of mRNA per ug total RNA, instead of moles
mRNA per mg total RNA.
Three lines seem to work the best for doing the standards for Tuesday's RNA gel, unless you are good at figuring out curves freehand (or at getting SigmaPlot or similar to fit a new equation to the line).
I will be available today until at least 6 or so, and possibly later depending on how my stomach is feeling (it has been queasy...). I will be in tomorrow (Ag Field Day, so Lipman's doors should be open) after 2:00 and possibly sometime before 2:00. I will be in most of Sunday.
4/26/07:
For the qPCR results, "Ct" is the number of cycles before the level of DNA
goes over the threshold.
In regard to the standards for at least Tuesday's gel for the RNA lab, there appear to be some headaches in the line, particularly at the lower end. The highest of your bands should be around 3375 bases; I would not use any standards heavier than 4 kb (4000 bases). I am not sure if the equation given in the lab manual will work; it is possible that using 3 straight lines will be preferable - I need to talk to Dr. Chase on this.
Note also that at least the Tuesday RNA gel perceptibly slants in how the dye front and bands travelled; you should use Rms, not cm, for it, dividing by the distance to the dye front for your lane and the standards - these numbers (distance to dye front for your lane and distance to dye front for the standards) are going to be different. The only exception would be if your lane happens to be right next to the standards.
For the RNA lab, the size of 25S rRNA for Arabidopsis is rather hard to find; it's 3375 bases. For the size of 18S and that of tRNA, see your general biochem textbook.
4/25/07:
For the qPCR lab: I have 1 copy of the B row group data, 3 copies of the F
row group data, and 3 copies of the C row group data. The A, D, and E row
group data have been given to a lab partner in that group who should give
them to the other people in the group.
If you haven't gotten to that stage yet on the RNA/qPCR lab, you do not need to subtract the picograms (or nanograms) from the no-template controls from the experimental values. It's fine if you've already done so, however. (We aren't sure whether it's a good idea or not...)
With regard to the RNA/qPCR gel, you only need to analyze your lane(s) and the standards, not the other lanes.
4/24/07:
In regard to plotting the standard curve for the RNA lab, you can do the y
axis as log(bp), not as bp on a logarithmic scale (you can do the latter if
you want, but I would not).
I will be posting up material on the RNA lab results for Tuesday later (Dr. Eveleigh's class then Dr. Ward's class got in the way). I will probably simply make copies to give to people of the results (both I and Dr. Chase would have copies), although it is possible I might scan in some of it and post it up on here.
To clarify: The material on this webpage regarding the sequencing lab replaces section 5; unless clearly stated otherwise, it only amplifies on other sections. This includes regarding "read a paper for extra credit" - while you can probably get a bit of extra credit by going into more detail about what one of the sequences that you find does, it has to be something that I can find out if it is right or wrong without reading the paper myself, which I do not really have time to do.
The idiots at NCBI have decided to rearrange the BLAST webpage; you can either use the old blast webpage or be sure you are using the right one ("nucleotide blast" == "blastn"). I also note that the idiots are having the BLASTN search (at least for the "old" blast webpage, and possibly the new one) default to searching the human database; use the "nr" database instead.
For the Tuesday lab, here are the listings of who is where on the RNA gel:
For the Wednesday lab, here are the listings of who is where on the RNA gel:
A picture of the RNA standards is at rna.std.gif (Invitrogen 15623-200); see below for the numbers.
Four things on the sequencing lab:
4/23/07:
The location of the final exam is room 106 of the Douglass Chemistry building
(aka the Heldrich Science Building).
I am grading; do not disturb (emailed queries may be answered).
4/22/07:
The sequencing labs are now due Wednesday (4/25/07) and Thursday (4/26/07),
for the Tuesday and Wednesday labs, respectively; I have not been able to go
as fast on the plasmid labs as I should have been, due to research needs. I
am attempting to have the plasmid labs (at least, those which were turned in
on time or not long thereafter, and for which there are no problems with
turnitin.com) available before the sequencing labs are due.
I am grading; do not disturb (emailed queries may be answered).
4/18/07:
The PCR labs are now due Thursday, 4/19/07; Laura will be grading them.
The RNA labs are now due:
4/17/07:
Note on the PCR lab in terms of getting data from other groups that the
Wednesday lab may have been using 0.5 mg/ml as the stock concentration, not
0.2 mg/ml.
The lab that would have been done today will be done next Tuesday. (For once, it'll be the Wednesday lab encountering any problems first, assuming they finish up the lab this week...)
I should be available most of the rest of the day today (I was dealing with, as noted below, the combination of war-on-some-drugs silliness and NJ's failure to have adequate drainage, not build in flood plains, etcetera (real good thinking given global warming, guys!)). I may post up further regarding when exactly I will be available until.
Hint on the PCR lab report, section THE REPORT, part 5: for why you might get less successful amplification (less amplification of template and thus less visibility of more-dilute bands) for longer sequences, try looking up "processivity" in regard to the DNA polymerase used for PCR.
Some stuff from last year's webpage on the PCR lab (I am not completely sure if all this is still applicable, not having read over things in detail nor talked with Dr. Chase about them...):
Note on the PCR lab, section THE REPORT, number 2: This asks for the amount of template DNA in the lane, which is not the same thing as the amount of template DNA that was in the PCR tube (you used 25 ul out of 50 ul in the PCR tube). The simplest way to calculate this is as a part of calculating for number 4 (see below), BTW.
For calculations for number 4, I have talked with Dr. Chase and the following is an acceptable alternative way to calculate the 4 numbers of molecules (one for each of your lanes) that you need to calculate (once for each lane):
In regard to the third part of question 2 on the PCR lab: "Why does more time not yield more product, i.e. the final value is fairly constant?" What this is asking is about the temperature the first part of the question was asking about - one of the three of 55, 72, and 94 - and why if you increase the amount of time for it beyond a certain point, you don't get any further increase in product. In other words, this is a temperature for which:
In regard to question 1 on the PCR lab, realize that, to get some tubes for which there is apparently no virus present, the solution must be very dilute (relative to the size of the samples). This is essentially a variety of "serial dilution" experiment like in microbiology. "Aliquot" means "sample", BTW.
4/16/07:
Note that there are no classes tomorrow (4/17/07); any labs due tomorrow
will be due Wednesday at the earliest.
I have not as yet been able to get in touch with Emilia on the standards for the RNA gels, so have not posted pictures of them up as yet.
My apologies that I have not been very available, but I am currently under some degree of stress for various reasons (dissertation research, running low on medication (accentuated by not being able to get to Princeton to my psychiatrist due to weather today - and, thanks to the DEA et al's War on (Some) Drugs, one of my prescriptions has to be gotten in person (no faxing, calling, or renewing...)), and other matters).
4/13/07:
I have noted several people who, instead of uploading to turnitin.com their
paragraph regarding how they put the restriction map together and determined
where the probe bound, put into that "assignment" the procedural writeup. I
have removed these assignments; you need to upload the correct information,
immediately. (I have also noted some cases in which people uploaded the
procedural writeup and the assembly paragraph, which is unfortunate
but OK.) In one case, the person seems to have instead uploaded the
procedural writeup into the "slot" for the answers to the questions, and
uploaded the answers to the questions into the "assignment" for the
restriction mapping paragraph. I have removed this person's procedural
writeup and moved the answers to the questions to that "slot"; that person
likewise needs to upload the paragraph regarding construction of the plasmid
ASAP.
The standards for the PCR lab are the same as for the plasmid lab (see below); the dye is tending to be at a lower kB than before (the 1018 band is, as far as I can tell, significantly above the dye front, although bands below 1636 are unfortunately very hard to see).
I need to check with Emilia for the info on the RNA lab standards, so that I can post up a picture of them and of the RNA lab gels.
I am not sure how much I will be available this weekend; I am trying to schedule a checkup on my transmission for Saturday, and am running low on one of my medications so may not be up to helping people as much as I might be. (I will not be available today (Friday) after 6:00 PM.) I also would like to try to start grading the plasmid labs, but that will depend on, first, how many of them I have so far, and, second, on my medication situation (the war on some drugs has resulted in one of my medications being rather hard to get - as in, I have to make an appointment with my psychiatrist every month for it, it cannot be refilled or prescribed over the phone...). The "assignment" for the PCR lab questions (or, rather, for the answers to the questions - only the answers, not the questions themselves!) is up on turnitin.com.
4/11/07:
I will try to post up the pictures of the RNA lab gels today or tomorrow, as
well as a picture of how the standards are supposed to look (we had some
stray bands in them on Tuesday, possibly on Wednesday).
In regard to the sequencing labs, note that where the BLASTX results have about the number and percentage of identical residues it calls it "Identities = #/# (#%)". What you want is the number in parentheses, which is the percent identity - but it never actually says "percent identity" on the results.
4/10/07:
I will be going home tonight after I take pictures of the RNA lab gel. I
realized that I went to sleep after 1:30AM and then woke up at 7:30AM, which
would explain why I am feeling groggy...
Note for the RNA lab for Tuesday that people in groups which did not do the cDNA this week will need to come in 1 hour early next week to do the cDNA. One of myself, Dr. Chase, or Emilia should be there.
The standards for the RNA lab are as follows:
In regard to the PCR lab:
4/9/07:
I am back in, BTW.
I have put a couple of updates in the info
(below) on the sequencing lab. I
woke up around 6 AM today; I am going home for a nap.
4/7/07:
Here are the PCR gel pictures (including one from Wed without initials);
sorry about not having them up earlier. If there are multiples, I advise
using the medium one unless we can do something about combining them. I have
done a bit of image manipulation on them, especially the two from wednesday.
4/6/07:
I woke up at 6:00AM today due to my apartment-mate's breakfast setting off
the smoke alarm. I will be available from ~4:30 PM (after the Fermentation
Seminar) till 6:00PM today. I will be available from sometime tomorrow
afternoon until around 6:00PM tomorrow (Saturday) likewise. I will be more
available on Sunday.
4/5/07:
I will be available most of the day today, except for some breaks (e.g.,
between 7 and 7:45); I will leave here at 10:00 PM or later. Note that I am
not much in a mood for dealing with people any more than is necessary... I
am not an extrovert, so having lots of people around asking questions does
qualify as somewhat stressful for me!
In regard to the PCR lab, I will try to copy the pictures from both days to this website today.
4/4/07:
I will be available this afternoon starting around 12:30 for help with the
plasmid labs. I do not think I will be able to stay as late tonight as I did
last night, due to sleep loss - besides, people, I was available Saturday
and Sunday and did not have very many people coming by, which is not my
fault...
On page 31, it asks for identification and sizes of the three forms of the plasmid (and of the RNA) from lane 1. These may not necessarily all show up in lane 1; particularly for the linear, they may show up in lane 2.
Anyplace the plasmid lab manual asks you to give the molecular weight of DNA or RNA - but not for proteins/amino acids, as in question 2! - it is asking for bp; you do not need to figure out the grams per mole of DNA or RNA.
4/3/07:
I will be available during the lab while the PCR and gels are running to go
over plasmid lab stuff. I will also be available after the lab, except that
I will probably need an hour or so of break-time after the lab finishes
up (as in, until 8:30 at least...). I intend on being here until after
midnight, although I will need some breaks and to go out at some point to
get some food.
The sequencing labs are now due the 24th/25th (Tuesday/Wednesday,
respectively)25th/26th of this month. This is partially
because they would otherwise have been due the same day as the PCR lab, and
partially because I doubt I will have the plasmid labs graded before then
(and I make no guarantees as to how long it will take me to grade labs that
are turned in late... a T grade is possible).
4/2/07:
Several things:
4/1/07:
Playing any variety of April Fool's joke on me (including involving me in
any variety of "Assassin"-type game) will result in severe grade-point
damage (as in, I will refuse to give you anything other than a 0 on labs
that I grade, my question(s) on the final, etcetera). It may also result in
bodily harm if my temper gets triggered, as it is likely to if, say, someone
squirts me with a water pistol around computers or electrophoresis equipment
(dangerous, people!).
I will be available today from 10:00AM till 5:00PM for plasmid lab assistance. I am not sure yet how much I will be available after 5:00PM - I will almost certainly take a break at 5:00 or so for at least an hour, possibly more, and I will probably go home tonight around 9 or 10 PM. (If family members wish to detain you for Palm Sunday services or whatever, show them this webpage...) If I am not in my office and I am available, try 202 or 207 (the extensions are the same as the room number, BTW) - I may be using the whiteboards to draw plasmid maps.
In regard to plasmid mapping, generally speaking the assembly is the hardest part - but before one can do the assembly, one has to work out the sizes of everything. I generally recommend using data from both the gel and the blot, including sizes from both (either by working out a conversion equation or, if you are lucky and the band Rfs from the gel and blot are about equal in all pairable bands (for "about equal", keep in mind that this is on a log scale so small differences blow up quickly!), just using the Rfs from the blot with the restriction ladder from the gel). I advise doing this for a couple of reasons:
In regard to the yield calculation, do not worry too much about the micrograms of DNA or nucleic acid; much more important is the concentration of DNA and of nucleic acid, and the percent DNA of the nucleic acid.
3/31/07:
In regard to the writeup of the plasmid lab (or sequencing lab, for the part
you did in 206/207) procedure, feel free (when summarizing it as I tell you
to do below) to refer to "step
17" or whatever, provided you are using the Spring 2007 lab
manual.
Sorry if anyone looked for me between 2:00PM-3:00PM (although I did say "something like" below...). I have not seen anyone so far today; I will try to be here until 9:00PM or so tonight.
I am told that there are some problems with logging into turnitin.com; I will try to check on these.
3/30/07:
Nobody was here yesterday for plasmid lab help; I have not received any
indication that people will be here today (as opposed to Sunday). I will
nevertheless be available 10:00AM-noon and something like
2:00PM-3:00PM. Note in terms of Sunday that I am definitely scheduling time
for Sunday afternoon; not sure about Sunday evening (as in, I do not know
how much I will be available after 5 or so); will probably be available at
some point (10:00AM or so, perhaps) Sunday morning.
I have decided to push back the due date for the sequencing lab by 1 week, since I doubt I will have the plasmid labs graded by the current due date. Therefore, the sequencing lab is now due April 17/18th.
3/28/07:
With regard to helping people with the plasmid lab, I will be available
Thursday starting around 10:00 AM, going until 3:00PM or so, then from
5:00PM or so until 9:00PM or so. I will post up times for Friday, etcetera
later. I may schedule a time Sunday afternoon where a lot of people can come
by and I can sketch up stuff on the whiteboards in 202 - it often helps to
take a look at the preliminary maps from plasmids from other groups, since
the plasmids are all differently-trimmed versions of the same thing.
3/27/07:
No lab lecture this morning.Actually, there was one, but
nothing required was covered.Remember to go to Foran Hall 124 (or
just to the lobby where we can meet) first during the lab session this
week!
With regard to turnitin.com for the plasmid lab, I will also be
putting have put up
an "assignment" for you to put in your paragraph or so (from the
plasmid lab) describing how you put the restriction map togther. (Yes, I
will be understanding given that it is sometimes hard to describe things
that are considerably pictures - but I do need to make sure that it is not
just one person (or two people) in a lab group doing the plasmid mapping
(that will also hurt you on the final, of course...).)
The below is the material from my webpage for last year for the sequencing lab (which I am grading); there may be modifications once I check with Dr. Chase as to any changes to his writeup: The below is in replacement of section 5 in the listing of THE REPORT (page 8) for the sequencing lab:
With regard to section 4 of the report and its question about whether you can see any vector sequence, you only have to answer regarding your own sequence. Answering regarding the entire classes' sequence is extra credit - not much extra credit, since all you really need to do is paste the entire sequence into the "VecScreen" program available via the BLAST webpage and see if anything pops up as significantly resembling your sequence. The best way - aside from VecScreen - to tell whether you may have any vector sequence is by taking a look at the results from the BLASTN search for your sequence. If the ends of your sequence are inside an existing sequence, namely Dr. Zylstra's, which doesn't have any vector sequence in it, then it can't have any vector sequence in it.
With regard to some of the questions in the sequencing lab:
For the sequencing lab, if you are unsure which file is which, the number from the tube was used as the first number after TC-. If you used another number's sample by mistake (because you looked at the second number, before the .ab1), and have already done the searches et al, don't worry about it. But everyone needs to be sure to make clear which sequence they did searches using.
3/26/07:
Remember to go to Foran Hall 124 (or just to the lobby where we can
meet) first during the lab session this week!
I have set up turnitin.com sections for the two lab sections; the numbers
are as follows (ask your respective TA(s) for the password; the wednesday
lab TAs should have it sometime today (I emailed them)):
People have asked about the final exam time; it should be 8:00-11:00 AM, Monday, May 7th, according to http://scheduling.rutgers.edu/springfinals.htm.
I will be definitely available for working on plasmid labs with people Thursday, Friday except Friday evening/night, Saturday, and Sunday. For other times (Friday evening/night is not available), please email to ask.
3/20/07:
The due date for the plasmid labs is now April 3rd/4th (for
Tuesday/Wednesday respectively). I have uploaded the pictures that I could
locate off of the computer in 206 of the gels (and a couple or so
blots). You should probably be mainly looking at the "med" (medium) pictures
below, except for areas which are too bright or dark for which you may need
the "low" or "high" ones, respectively. I may find the time to work on
combining these, especially if people are having problems with a particular
gel in this regard. The ones that I have are:
My car is in the shop, which is limiting my time to some degree; I should have it back Friday - hopefully!
Note that I normally give a sequencing question on the final exam (I will
try to remember to check on exactly when it is), and plan on doing so
this year. (A plasmid mapping question may be from me or may be from Dr. Chase,
but is also likely in any event.) I have put an example of the sequencing
question I generally ask in
expbio.question.example.txt; it
also includes the grading master (this is the same example from last year;
I may put up a different version as well). Note that the sequence in the
question (and thus the master) varies from person to person, and that the
question itself might vary from the example (probably only slightly, though,
such as in the exact points for everything). Note also (I will make this
clear in the question itself) that 32P in dNTPs does not mean
that there is 32P in the primers.
3/18/07:
Due to car problems (probably transmission - groan!), I will not be
available on Monday, 3/19/07. I will consider pushing back the due date for
the plasmid lab further.
3/9/07:
I will be doing image manipulation on and posting up gel and blot pictures
when I get around to getting them off the FotoDyne computer (for gels) or
the computer with the scanner (for blots), or when I am given them in
person (emailing them will not work!). So far:
If, for your blot, you were able to see and mark the position of the dye front, I advise figuring Rfs in terms of (distance from well to band)/(distance from well to dye front), not (distance from well to band)/(distance to end of gel). This is particularly needed when the dye front varies in distance (although admittedly this will be hard to see on the blot, and the dye front tends to be large enough that the curvature is hard to quantify - halfway between the top and bottom of the dye front can work well).
The Standards for the plasmid lab:
The brightest band should be the 1636 band; the 506/517 band should be the second-brightest after that, unless it's covered over by the dye marker.
3/8/07:
In regard to my schedule, I am planning on being in until late on Friday. I
will not be available on Saturday and will not be here most of the time
(especially after 3 PM). I should be available for turning in labs
only - maybe for emailed questions, but these may
also take until after spring break! - on Sunday. I am not sure about the
rest of the week and the following weekend, but at the most will be
available for turning labs in and maybe emailed
questions (keep in mind that I cannot read attachments!), not for anything
else in person, until Monday the 19th - and I may not be available for
turning labs in unless you specifically check with me and make sure, or I
announce it here.
I am hearing different information on exactly when the lipid labs are due, what bonus points are available, etcetera. I will post up when I hear from Dr. Chase. BTW, another link that may be of assistance for the lipid labs is www.lipidlibrary.co.uk.
For the procedural writeup on the plasmid lab, I just want:
3/6/07:
There is no material to be turned in on turnitin.com for the lipid or
carotenoid labs. The first lab that will need to be turned in on
turnitin.com will be the plasmid lab; I will create the class and distribute
information on how to sign up for it sometime prior to the plasmid lab being
due.
Please keep in mind that I am not an organic chemist; I am a biologist, albeit one with considerable exposure to biochemistry by this point. Most questions on the lipid lab, particularly any having to do with the interpretation of plates (or most other things than those having to do with the computers) are better directed to Dr. VanEs (or, if it is a question on how the report should be done, to Laura, since she will be grading the lipid labs); similarly, most questions on the carotenoid lab, again except for those having to do with the computers, are better directed to Dr. Chase.
3/5/07:
REMEMBER TO DO THE PREHYBRIDIZATION (today for Tuesday lab
students, tomorrow for Wednesday lab students)! Note that we should be
getting the probe (needed for the second portion of the prehybridization)
sometime today between 1 and 2 PM. Emilia (thanks!) will be putting in
the probe for people in the Tuesday lab, due to its late arrival (it will
not be available until at least 3:30PM), if you do the first
portion of the prehybridization before she leaves (which is generally around
4:30PM or 5:00PM).
The due date for the plasmid lab, which I will be grading, will be no earlier than the 27th or 28th of March (for Tuesday/Wednesday), partially because I will not be available for most of spring break (any of it except for the 9th of March (this Friday), by my current plans).
The due dates for the lipid and carotenoid lab are as follows:
I will be here today until sometime well after 5 (probably at least until 9, although I got to sleep last night at around 3 and woke up at 8:30, so I am not sure about that), such as to help people with the prehybridization and with what parts of the lipid and carotenoid labs I can help them with (please do not ask me things on the plates, for instance - ask Dr. VanEs on the lipid lab, Dr. Chase on the carotenoid lab).
3/2/07:
For the phosphate determination and similar things with the Lipid lab,
if you have at least 3 different amounts of samples (aliquots) taken from one
base solution (e.g., for the phosphate determination, something like 10, 20,
50 ul of the phospholipid solution), then you can plot the resulting
absorbances on the Y axis vs the amounts on the X axis (plus a 0,0 point
unless something went wrong with your blank), take the slope of the
resulting line, and divide said slope by the slope of your standard curve
(which should be something like absorbance (Y axis) vs uMol (X axis) -
convert mls or uls of standard into molarity, e.g., uMol), to
get concentration (in the example, uMol/ul). You can then take that and
multiply it by the total amount you had of the base solution to get the
amount of the base solution. You do not have to do it by calculating out the
amount for each and every aliquot if you do it this way.
I should be available a lot of the time on this Saturday and Sunday; email to make sure of any particular time.
3/1/07:
In regard to interpretation of the GC results, Laura found the following
link which may be of use: http://www.midi-inc.com/media/pdfs/Fatty_Acid_Profiling.pdf.
A local copy is at Fatty_Acid_Profiling.pdf.
2/26/07:
Emilia now has GC files for all (lipid) groups; see her for them.
2/23/07:
The HPLC files are now (also) on the computer in 214 closest to the window,
which has the program to view them (Chemstation). They are in the
directory "Chase2007Carotenoids".
2/22/07:
The test gel for the Tuesday lab is available at test.gel.plasmid.lab.tif; the ordering
of groups (from left to right) is as follows:
I should be able to transfer the HPLC files from the HPLC computer to one (or more) in 214 tonight or tomorrow; the due date for the Carotenoid labs is accordingly 2 weeks from tomorrow (as in on the 9th of March). If Emilia has been able to get all the GC results to people, then the due date of the Lipid lab will be two weeks from whenever that took place (at the very latest, 2 weeks from tomorrow, the same due date as the Carotenoid labs).
12/20/06:
I have now graded a total of 53 labs. If you have not put in the
electrophoresis lab in turnitin.com, then I have not graded your
lab, and you may be getting a (low) T grade. The mean is now 95; the median
is now 97; the min is now 68; the high is still 109.
12/18/06:
I have most (43 at last count) of the electrophoresis labs graded. I will
bring the ones that I have finished with me to the final exam for
experimental. The remaining ones either were turned in late or have some
other problem. The grades so far are overall very good (mean/median 97/98,
low 71, high 109), but this may change as I grade the remaining ones (I
would anticipate, from past experience and other information, that some of
the remaining ones will be good, but a higher proportion will be bad than is
the case with the ones I have graded thus far).
12/17/06:
To clarify one misunderstanding: Yes, you are allowed to use graphing
calculators on the exam. You are not required to use such a
calculator.
12/16/06:
Explanation from Liora (edited by me) - thanks very much to her! - for the
slope-point method of getting the equation of a line without a computer or a
graphing calculator: slope.point.explanation.txt
The exam for Experimental is, as far as I know, in the Ruth Adams lecture room (basement - 001, I believe). Except maybe via email (and do not ask when your lab will be graded!), I am not available except for those who need to hand in a lab.
Places to check for how to get equations of lines with a TI-83 Plus (may work with TI-84/TI-84 Plus/TI-83 Silver Edition):
12/14/06:
As noted below, I will not be available today until at least 4 or 5 PM. I
should then be available until quite late - at least midnight, probably
later (up to 4 AM or so).
12/13/06:
MMP blot, further versions:
I am finding that, if you have a very blurry blot, using TotalLab on it (even though you are not required to) is sometimes helpful. However, it is usually preferable on blots on TotalLab to set the removal of background via the "rolling ball" to 200, not the usual (for gels) 75.
I will be in tonight until sometime after midnight (barring running out to get something to eat or run other brief errands). How late will depend on how late people are here. I will be somewhat available tomorrow for further assistance, primarily in the evening/night (there is a holiday party at noon on Thursday and I may consume some alcohol...). Friday afternoon, I will start grading and will not be available for anything except turning labs in - I will have up a "DO NOT DISTURB" sign and anyone bothering me (except, as I said, for turning labs in) is:
Hint for question 1: Think about what you would see if you ran hemoglobin on a SDS gel then stained with Coomassie vs running hemoglobin on a native gel then stained with Coomassie.
12/12/06:
MMP SDS Blot: Fri.MMP.SDS.blot.tif
and an edited version: Fri.MMP.SDS.blot.edited.tif
In terms of the pictures that you need to present, the important things are indicating which lanes are which and marking where you measured to. You can put the identification of bands, including the standards, in tables instead of on the pictures, if you prefer.
Revised version of AMO native blot: Native_AMO_600dpi.tif.
Rf and Rm are the same thing, by the way.
In regard to the isoelectric focusing lab:
A few notes on how the standards are looking:
It is generally easiest to calculate molecular weights and pIs if you put the Rm (or distance) on the X axis and the log(mw) or pI on the Y axis. Then, you can simply plug the Rm (or distance) into the equation of the line in place of x, and get y, which is your log(mw) or pI, depending on what you were graphing.
12/11/06:
In terms of the lab being due Wednesday, that is indeed for all lab
sections, and that is also indeed for both the Electrophoresis and the
Isoelectric Focusing parts of the lab report.
Note: You do not need to plot the D-amino-acid-oxidase (abbreviated DAAO) band on any of your graphs, despite what it says in the lab manual, unless you have decided to do a manual plot of Rm vs log molecular weight, probably using graph paper; you need to plug in the DAAO Rm into the equation of the line, get out log(molecular weight), take the antilog, and get molecular weight, and show both the result and an example of the calculations. (The only thing plotted on the graph should be the standards, in other words. But do not worry if you have already done the graph and you put a point for DAAO on it, since that is what the lab manual asks for.) On each table with the - or a, if you have isozymes - DAAO band on it, you should indicate which band it is.
TotalLab is now installed on the next-to-last row of computers in 202, thanks to Peter Anderson (as in the PCs - Dells, I think - in the last row that is facing forward), as well as in 214 and on one computer in the very last row of 202. However, you may not be able to do the "Export to Excel" on the computers in the next-to-last-row of 202; do "Export to File" instead and set it for ".csv" (Comma Separated Values) format. That can be read by either Excel or by OpenOffice Calc, which is on the computers in the last row of 202.
The gels we used had two sections: a "stacking" gel and a "running" gel. These differed in concentration, such that the proteins "stack up" when they run into the interface between them. For some people, the stacking gel was cut off along with the wells. Others do not seem to have done this; for them, they need to be measuring from the top of the running gel to the bands and dye front, not the top of the stacking gel. It appears that the distinction between the stacking and running gel is visible on the coomassie-stained SDS gels by that the stacking gel took up more dye. The proportion of this to the entire length of the gel should then be the same for the corresponding blot.
The TotalLab directions are assuming that you are on one particular computer; when you are directed to save things in a Class413 directory or anything like that, just save it on the desktop or wherever you have the image file you are working with.
A further edited version of the NKSP1 blot: NKSP1.western.blot.edited1.tif The colors of the prestained standards for that blot: Blue, Magenta, Green, Violet, Orange, Red.
The MMP gel (which turns out to have been under "Katz" below...): Fri.MMP.tif.
Two combined-exposure versions of a Tuesday SDS blot:
Two edited Friday lab gels:
An edited Tuesday lab Native blot (Michael/Jen/Clifton):
In regard to TotalLab directions at step 12, do not click on the arrow pointing right and saying "Next"; click on the arrow beside that, that is pointing down, and select "Finish".
Here are the Wednesday IEF gel pictures:
Note for the Wednesday and Friday IEF gels that the two small marks at each side away from the tab end (at the 4.0 pI end) appear to mark where the 4.0 pI electrode was.
12/10/06:
Another edited Tuesday SDS gel - this one unfortunately seems to lack
(non-prestained) standards: Tues.Rajni.Jen.SDS.edited.tif
And an edited image of one wednesday SDS blot: NKSP1.western.blot.tif
12/9/06:
Four things on the questions:
Note that you may need to distinguish, in the pictures of your gels, where the top of the running gel is; the stacking gel, above it, seems to pick up more stain in some assays, so may be visible by an increase in the background. You should be measuring from the top of the running gel. If you cannot distinguish where it is, please ask me about it - I should either be able to see it or able to construct an equation to figure it out.
Note in regard to downloading images for use in TotalLab that you should do so using Firefox/Mozilla, not Internet Explorer (aka Internet Exploder...); it gets messed up if you use Internet Explorer (or if you open the file instead of right-clicking to save it).
Here are some more Friday blot images:
And some combined-exposure Tuesday gel images:
12/8/06:
The Electrophoresis/Isoelectric Focusing lab report (it is one report, even
though the material is in two different packets) is now due Wednesday,
December 13th, not Tuesday, December 12th, due to people having final exams
on Tuesday, December 12th. (This is for all sections!) However, this means
that I am highly unlikely to be able to get back them to people prior to the
exam (I had some doubts as to whether I would be able to anyway, but this
makes it pretty certain...). Hopefully, I will be able to do so at
the final, but no guarantees, especially for any that are in later than
Wednesday!
Here is a picture of the Tuesday IEF gel (coomassie version) with standards marked: Tues.IEF.Coomassie.standards.tif. As you will see, the line for the electrode at the end away from the tab (wedge-shaped thing) is a pI of 4.0; the line at the tab end is a pI of 6.5; the rest should give you a reasonable line. I will try to remember to create a Friday section version of this (and will do the same with the Wednesday section ones when someone gives me a copy of the pictures).
Friday pictures of blots (do look at all the file versions!); not all of them are up as yet:
Isoelectric gels for Friday:
Other friday pictures I have gotten today:
In terms of what is required for TotalLab vs manual measurements:
12/7/06:
Here are the Wednesday pictures that I located from the scanner in 214 (note
that I moved them, and the TotalLab files that were associated with some of
them, to a Wed.Gels directory (which Microsloth
calls a "folder") in "My Documents"):
I should be available this evening, most of Friday afternoon and evening, and most of Saturday and Sunday for working on gels. You do need to do TotalLab analyses on the SDS gel results, in order to be able to compare the amounts in the bands for the acid supernatant sample with those for the final enzyme sample. You can generally do either TotalLab or manual analysis of the native gels, except that if your results show isozymes (more than one band in a given lane showing activity), you will need to use TotalLab to see what percentages are in each of these bands; you will get extra credit for this extra work, however.
Here also are some edited pictures from the Friday lab gels:
And Wednesday:
And an edited version of one Tuesday blot: Native_MCJ_600dpi.edited.jpg
Note in regard to TotalLab and this website: When downloading pictures for processing in TotalLab, do not click on them to display them then save them from the graphics display program; right-click on them then hit "save link as" using Firefox (Mozilla) or equivalent.
12/6/06:
Here are more blot pictures I have been able to locate for Wednesday; I will
be checking the computer in 214 with the scanner sometime tomorrow (someone
was using it when I checked, with no other computers being free):
12/5/06:
With regard to the final for Experimental Biochemistry, I recommend looking
at old exams (see http://www.aesop.rutgers.edu/~dbm/tedchase.html
for most/all of the ones that are available;
IRIS (library system) may have
some more material available under "Reserves", however, but I have
not checked). In addition to studying these, the introductions to the labs,
and the procedures that you did to work up the data for the labs, I
recommend also looking at the example version below of the question I plan on doing for the exam.
Here are the gel pictures that I have so far; these are mostly unedited, so are likely to be improvable by photomanipulation (and I will try, if I can find the time, to do said photomanipulation on them, but you are encouraged to try doing so also):
In regard to my schedule, I will be available this evening until midnight or so; I should be available tomorrow (wednesday) and thursday evenings likewise (during the days I need to do things like renewing my driver's license); I should be available most of the day and evening, possibly some of the night for Friday/Saturday/Sunday, except that if I get to bed late enough, I will not be available in the mornings. If you want/need to come in very late (including the next morning very early...), email me to let me know, or if there are no students here I may go home earlier...
12/3/06:
BTW, if you are calling me on the phone, please keep it brief/short - I
really hate talking on the phone...
12/2/06:
People who have turned in enzyme lab reports: Be sure you also put in the
questions and procedure to turnitin.com! Not everyone has done both (and
yes, I checked to see if people had combined them, which at least one person
has done, unfortunately...).
For the final exam for Experimental, I will be doing a section on how to do a procedural table and how to do a purification table, the numbers for which will differ for each person. At least part of it will look like the following linked file: fall.example.question.answer.txt; I am not sure yet whether I will add anything else to it.
I will be in this weekend in the evenings and nights, possibly in the afternoons, depending on what time I get to sleep in the mornings. I can accept lab reports and give various assistance with them.
11/29/06:
Note that, as far as I can tell from http://scheduling.rutgers.edu/fallfinals.htm,
the exam for Experimental is Monday, Dec 18th from 12-3:00PM and the exam
for General Biochemistry is Wednesday, Dec 20th from 12-3:00PM. Please let
Dr. Chase or Dr. Kahn, respectively, know well before the exam if you have a
conflict.
Reminder: You also need to submit the procedural writeup and the answers to the questions, in separate files, to turnitin.com. You should remove everything but the text of the procedure and the text of the answers to the questions (do not include the questions themselves) before you upload these files. (E.g., do not include graphs or tables!)
In regard to how the account of purification and the data tables+graphs for the Lowry and enzyme activity (e.g., pyruvate) assays should be arranged (the below is a quote from an email last year from him, replying to one from me):
Allen Smith wrote:
P.S. BTW, in the section "The Report", you ask for assay and protein
concentration data in tables and graphs in the account of
purification. Then you say to give a partial description of the experiment
(assay?) procedure, then graphs, then explain the data. Is all this
interleaved in with the account of purification, including descriptions of
how to do the assays, or is it after the account of purification? And after
that come the experiments (as opposed to the assays)?
Dr. Chase wrote:
On paper I could take it either way; indeed, the purification may be
easier to follow if the data are interweaved with the account. But
they should save a data-free Account of Purification to send to
turnitin.com.
In the interest of not expending massive quantities of paper, particularly if it is from one of the Lipman Hall printers, I personally would cluster the tables+graphs together as much as possible, since otherwise one has to either:
11/28/06:
I should be here until at least 11:00 PM tonight, probably until after
midnight, and probably will be in until at least 10-11 PM tomorrow
(Wednesday) night, possibly later. Do not knock on any windows
other than those associated with my office (near Chang end of building, not
near Loree end of building) to get let in and do not prop any
exterior doors open. Call a friend who is already inside, call me, or knock
on my window, in order of preference.
11/27/06:
In regard to the peroxidase assay, when it talks in the lab manual about
using the highest slope, that is referring to the highest slope in a given
run - only applicable if you used the Cary to get the graph and the line was
curving.
11/25/06:
I can answer some questions via email this weekend, but replies may be
delayed. Email to ask if you think you need to come in for whether I'll be
available.
11/22/06:
It is 10:05 PM, and all the students working on labs here have left; I will
be going home sometime a bit after 10:20 PM, unless someone emails/calls me
that they are rushing here with a lab to turn in...
Note on question 2: You can work it out in terms of ml stock enzyme (as in you do not have to convert it to micrograms), as long as you are clear that that is what you are doing (and you make sure it is ml stock enzyme - allowing for any dilution factors!). Do not give an answer of how to get a more sensitive assay of "concentrate the enzyme more", however!
Reminder from Dr. Chase: You need to label each axis in your graphs appropriately! "Column 1" and "Column 2" are not appropriate, and will get points taken off. I (Allen) would also add that if you are plotting something vs ml of enzyme, you need to state if it is vs ml stock (undiluted) enzyme, as in "ml stock enzyme", or vs "ml diluted enzyme". Simply putting on a page with the graph the dilution factor is not sufficient.
I will be here until at least 10:00 PM or so - probably later, if people are here working on their labs still. Labs turned in to me before I leave will be counted as in today, as usual; I will be dropping them off with Dr. Chase.
11/21/06:
Error on my part in regard to the polarograph! I am afraid
that, in order to get chart widths, one should divide the number of small
chart boxes across by 50, not 100 - there are a total of 50 of them across,
not 100! I apologize on this, and am informing Dr. Chase about this error on
my part.
I have created a rough draft of instructions on how to use SigmaPlot to do Michaelis-Menten fitting; it is available at sigmaplot.MM.txt.
Note from Dr. Chase: You need to label each axis on your graphs; he is seeing some carbohydrate mutarotation labs without labels on each axis!
11/16/06:
I will try to go over polarograph stuff 2-3 PM on Friday; look for me either
in my office or in the room beside the lab (207?).
11/15/06:
Well, going over the polarograph results did not happen yesterday - too
frantic (please calculate everything beforehand as much as possible!) - and
I doubt it will happen today or Friday. For anyone who is unsure on how to
interpret the polarograph data, please let me know when you can come by and
I will try to schedule a time when as many people as possible can do so and
observe the process (so neither Dr. Chase nor myself have to answer the same
questions over and over again!).
11/14/06:
I think that I will go over how to interpret the polarograph results
during the electrophoresis today; I will try to come by Wednesday and Friday
also, in case Dr. Chase is not available to do that.
11/13/06:
I may try to arrange sometime at which multiple people can come by and
get instruction on how to interpret the
polarograph results, so that neither I nor Dr. Chase are having to repeat it
over and over again. I believe that we will not
have lab the week of Thanksgiving break, so Tuesday afternoon that week may be a possibility - let me know if you are
needing this, please.
Remember that you can remove points on graphs if you have reason to, such as if the absorbances are going down with increasing amounts of enzyme, provided that:
In figuring out whether your protein mg/ml values make sense for the purification table, I suggest making an additional column on said table, of "total protein (mg)"; to get this, multiply your volume by your mg/ml. This should always remain the same or go down - at most stages, it should go down. If it goes up (significantly - as in not within error), then there is likely to be a problem.
One way to tell whether you need to remove the 0,0 point, which does happen sometimes in various assays/plots, is to look at the standard error of the Y intercept. If it is greater than the absolute value of the Y intercept, then that is an indication that you may need to remove the 0,0 point. It is not an absolutely certain way to tell, however; you have to use your judgement - consult us if unsure!
Several more things on the enzyme lab, copied from earlier years:
Also copied from earlier years:
Regarding experiment 5 and similar questions involving Michaelis-Menten plots of substrate vs rate:
One thing to keep in mind when calculating substrate (D-alanine) concentration for Michaelis-Menten and related graphs (e.g., Hanes-Woolf) is that this is the concentration in the reaction. 0.06 M DL-Alanine = 0.03 M D-Alanine; 0.03 * an amount in mL gives you mM (millimoles), but the (pyruvate assay) reaction is occuring in 1 mL volume, so you divide mM by 1 mL and that gives you back Molar. You can make this number more convenient by multiplying by 1000, provided that you then label [D-alanine] as something like [D-alanine] (mM), since [D-alanine] would normally mean molarity of D-alanine, not milli-molarity of D-alanine.
Note for figuring out the molar extinction coefficient for experiment 2 that you need to allow for any dilution used to get the spectrum. Divide the protein concentration (in mg/mL == g/L) by the dilution factor before converting it into molarity (moles/liter). Similarly, if you used a 2 mm cuvette so did not have to do a dilution, remember to multiply the absorbance by 5.
Note that if you did experiment 8 as well as experiment 5, you may want to use a Lineweaver-Burke plot to get the initial values for a Michaelis-Menten plot for both experiment 5 and experiment 8, since Dr. Chase wants you to graph using Lineweaver-Burke (and Michaelis-Menten, possibly - this is unclear; you definitely need to do a Michaelis-Menten plot and curve fit for experiment 5) for experiment 8 and include a line for experiment 5 on that graph. This would be instead of doing a Hanes-Woolf plot.
I am seeing a problem with a few people's graphs that were done on Excel:
11/11/06:
For determining activity by the normal pyruvate assay (as in not anything in
one of the experiments in which you do not vary the amount of enzyme):
11/9/06:
I will be in this weekend most of the time - afternoons and evenings unless
I am out running an errand, and possibly mornings and nights depending on
how my sleep cycle behaves. I should be available definitely for turning in
carbohydrate statistics and will try to be available for helping with
carbohydrate statistics and with starting on analyzing and writing up the
enzyme lab (which I suggest working on ASAP! You will need to have some
numbers for activity by your lab section next week at the
minimum...).
Dr. Chase says that the best thing to call the sample from before the final column is "Second ammonium sulfate precipitate". The stages to put on the purification table are then:
11/3/06:
I have modified slightly my SigmaPlot instructions for the mutarotation
analysis (at http://www.drallensmith.org/teaching/mutarotation.sigmaplot.txt);
the alterations are marked in brackets ([]). Some of the information may be
of interest to people using Kalidagraph, BTW. I will be in this weekend most
of the time (most - barring going out to run errands - time in the
afternoons and evenings at the minimum; I am not sure when in the morning I
will be in or when at night I will leave, so make an appointment with me if
you need me to be available at a particular time (I will try to fulfill such
time requests if at all possible)).
11/2/06:
Neither Dr. Chase nor Dr. Kahn have gotten back to me on the SigmaPlot
mutarotation analysis, so I will simply give you what I have available -
hopefully when Dr. Chase gets around to looking at it he will not have any
significant changes. It is at http://www.drallensmith.org/teaching/mutarotation.sigmaplot.txt.
People may find the discussion on my 2005-2006 webpage regarding Kinetics and Hanes-Woolf plots useful in studying for the exam, incidentally (and you will need to know about said plots for doing the enzyme lab data analysis; my apologies for not thinking to put this link up prior to the Kinetics problem set being due...); you might also want to look at http://orion1.paisley.ac.uk/Kinetics/contents.html, both for the exam and for the enzyme lab writeup.
10/30/06:
The carbohydrate statistical workup (mutarotation) is due two weeks from
when you got the sheet from Dr. Chase on what you are expected to do; this
should have been during your lab last week. I will try to put up something
on how to do the mutarotation analysis using SigmaPlot - I have asked
Dr. Chase (and Dr. Kahn) to take a look at what I have written up
currently (see the 2005-2006 webpage if you want to take a look, keeping in
mind that it may change!).
10/27/06:
I am not available this weekend, in general - I will be (tonight (Friday),
Saturday morning and afternoon, and Sunday all day) working on writing up a
rough draft of the introduction and literature review section of my
dissertation (Dr. Kahn wants it on Monday). Questions via email may be
considerably delayed in when I answer them! I can accept lab reports, and
can - if need be - let you into the building in order to accept them (or to
use the computers upstairs to do them - note that for the statistical workup
for the carbohydrate lab (mutarotation) you will need to use SigmaPlot or
KalidaGraph, not Excel, and so far as I know these are only available in
this building unless you manage to download a trial copy); I would prefer it
if you would get into the building via having a friend who is already here
let you in, however, since that will distract me less. Note on the
carbohydrate questions that the one on the best method to measure the
hydrolysis of a couple of different disaccharides actually is intended to
get two different methods as answers, unlike what I had thought earlier
(although what I had hinted to people to use is at least a partial answer
to the question) - errors like this on my part are a large part of why I am
reluctant to help people much with the carbohydrate labs! If at all
possible, please check with Dr. Chase or Dr. VanEs or (for questions
regarding what is wanted in the report) Laura for help with said lab.
10/23/06:
Remember to come in and do the dialysis! This should be
today for anyone in the Tuesday lab - preferably, come in sometime
between now and 3:30, so that Emilia can help you with it. I will be
available tonight after Emilia leaves, but I am not sure for how long - my
sleep cycle is getting weirder... plus I am still trying to work on my
dissertation writeup.
One mistake on my part on the carbohydrate labs: You are not required by the lab manual to graph the unknowns, as far as I can tell on a recheck. Sorry! And do not graph the unknowns on the graphs with the standards - that would not make any sense, especially since you would be graphing ml for the unknowns and mg for the standards.
10/21/06:
I am not available this weekend, in general - I will be (tonight and all of
tomorrow) working on writing up a rough draft of the introduction and
literature review section of my dissertation. Questions via email may be
considerably delayed in when I answer them! One frequent problem that I am
seeing with the carbohydrate lab is that people are plotting the results for
multiple sugars on one graph - you should only be plotting the sugars
(generally one sugar plus the unknown for most methods) that
varied in amount, not the ones that we only used one amount of. And unless
the lab specifically says otherwise, you should not plot more than one sugar
(including the unknown) on any given graph. Note that
carbohydrates are generally not my area, except when in nucleic acids, and I
will refer you to Dr. Chase or Dr. VanEs or (for questions regarding what is
wanted in the report) Laura for help with said lab - that the next lab due
is the carbohydrate lab, on which I cannot help much, is one reason I have
scheduled this weekend for working on my dissertation writeup.
10/18/06:
Several things (some important for all students in the course!):
For how to get rate (v, units/ml, whatever) for a pyruvate assay when you have too few points to use the slope method (e.g., when you are varying something other than the amount of enzyme):
10/11/06:
We have found another typo on page 3 of the statistical analysis handout
(statistical appendix for the protein lab) - up above, it says the standard
error of the mean for the example is 0.5686, while down below, it uses a
value of 0.586 (divided by the mean to get the proportional error of the
mean). This is not large but can be confusing.
10/10/06:
With regard to part 5 of the statistical analysis for the protein lab:
Note that the calculation for putting together the standard errors of the standard curve with the standard error of the mean (that you calculated last time) is the one on page 3, not the earlier version (with mg*square root symbol) on the prior page - the prior page version is given as an example and an explanation of how we worked out the method. Also note that there is a typo on page 3 for the example calculation of the standard error of the slope - the result of squaring 0.017024 should be (approximately) 0.0002898165, not what is on the sheet.
I will try to be available tonight for helping people with the statistical analysis, starting 45 minutes or so after the lab finishes up. I am not sure how late I will be able to stay - despite going to bed last night after 1 AM (and not feeling sleepy until at least midnight), I woke up before 8 AM this morning and was not able to get back to sleep... sigh!
10/8/06:
Please note that the Excel instructions below
are not specific to our class, but are general - you probably do not need
to do the "Line Fit" plot, for instance. Also note that, for the
Data Analysis option to display, you cannot have selected something on the
graph - indeed, the Excel Analysis Add-In will not actually do anything with
the graph; you need to have the line (and equation and R-squared value) on
there through the Add Trendline option (via right-clicking on a
point). (Excel is from Microsloth; what do you
expect, functionality without having to jump through hoops? Do not be so
silly...)
10/7/06:
For people who still have their absorbances but now lack the number of mls
for each tube due to turning in the datasheets:
10/2/06:
Remember that all of the methods except for the Coomassie Blue will have a
0,0 point, on both the standard curve graph and the unknown graph. The
Coomassie Blue will have a 0 mg protein point that will be above 0 for both
A595/A466 and A595. You will
then copy these points from the standard curves to the unknown curves, since
0 mg ovalbumin = 0 ml unknown protein. (No protein is no protein!)
You do not need to plot the absorbances of the unknown substances or of the interfering substances. You will get a rather weird graph if you do!
You can remove points if you have reason to, such as if the absorbances are going down with increasing amounts of protein (however, this is normal for the Coomassie A466), provided that:
For the Coomassie Blue, it is often best to create two standard curves - one not including points with A595/A466 above 2, and the other including such points (and for which you may wish to take out some of the lowest points - the first point after the 0-protein one is particularly likely to have a A595 value that actually goes down a bit, in which case it should probably be removed). Unknown protein samples with a A595/A466 below 2 should be interpreted using the first standard curve; others should be interpreted using the second standard curve. When you work up the statistical analysis, you may find the A595 will work best in some regions of the curve, but for now, just worry about the A595/A466.
For anyone (still) wondering how to get the E1 mg/mL for the protein determination procedure, here is the easiest way I know of to do it: For the methods in which you have plotted (for the standard curve) the absorbance vs the mg of ovalbumin, take the slope (which is A/mg) and multiply it by the number of mLs of solution used. (For the E1 mg/mL determination, this should be the total number of mLs of solution prior to taking out 3 mLs to put in the cuvette - e.g., 5 mL for the Biuret method. For figuring up sensitivity, it can be argued that one should instead use the total number of mLs of sample that you can use in a given method; we will be accepting either way of doing it.)
Several things on questions 1 and 2:
Some, perhaps all of your pH labs should be available during the Friday lab section - Laura, who is normally the Wednesday TA and is grading/has graded them, is taking the Friday lab this week, due to Rob (the normal Friday TA) recuperating from (relatively minor but rather painful) surgery.
9/19/06:
Two things on the questions for the pH lab:
9/11/06:
I have (as of 9/8/06, actually) set up the Tuesday, Wednesday, and Friday
turnitin.com sections, and have sent the ID #s and passwords for them to the
TAs for the Wednesday (Laura Youngster) and Friday (Rob Shortell)
sections. As per the below, the section/class ID number for the Tuesday
section is 1612348; email me for the password.
9/6/06:
You do not need to submit anything from the statistical calculations (due
next week) to turnitin.com. I will see about getting the turnitin.com
class ID number and password for the 3 sections sometime today
(Wednesday).
As instructed in the pH lab manual (pages 4 (top/middle of the page) and 12 (bottom of the page)), there are two calculations that you are supposed to do ahead of time. I will be counting whether or not you do so as a quiz. Note that I will not be counting whether you get these calculations right, however, since you are allowed to work together on them (and indeed are encouraged to do so if you have any problems), unlike on a quiz - but I will be checking to make sure that each person has at least tried to do said calculations, and counting this as part of your quiz grade for the semester.
There is now a Wednesday lab experimental biochemistry page, incidentally, albeit somewhat blank at the moment - see http://myspace.com/experimentalbiochemistry.
I have created the turnitin.com account for the class as a whole and for the Tuesday section, and will be creating the accounts for the other sections once I make sure what email address the other two TAs want to use for dealing with turnitin.com. For the Tuesday section, the class ID is 1612348; to send you the password for joining it, I will need your email - as requested in the lab, we need each person in the Tuesday section to email me (see below for the address, making sure to read the instructions fully and carefully!) from whatever account you wish to use for correspondence with us. The first thing that you have due that needs to be put up on turnitin.com are the pH lab questions, which are due one week from your lab period next week (you will be doing the pH lab next week).
9/5/06:
Dr. Chase sometimes mentions "prelabs", and sometimes other TAs do them. I
don't. However, I will be treating the two calculations for the pH lab
(starting next week) that you are supposed to do before the lab period (see
the lab manual) as a "take-home quiz" of sorts. You are allowed to work
with your lab partners on said calculations, however, unlike a normal quiz,
so all I will be counting is whether you tried to do each
calculation, not whether you got it exactly right.
People frequently say that the lab takes more time than it should for the number of credits. You are correct; Dr. Chase and I agree with you. Unfortunately, it appears to be University policy, probably due to the various humanities departments lobbying, to not count laboratory hours as much as classroom hours - even if the laboratory in question has, like Experimental Biochemistry, lab reports that require lots of time outside of class. On the other hand, do realize that this is one of the more thorough biochemistry (and related areas) laboratory courses that undergraduates might ever take, and definitely gives people lots of experience - experience that has meant the difference between getting a job and not getting a job for some. (The course credit hours have been increased a bit via adding extra time to the lecture, plus combining the course with Data Treatment, but it's still too few credits in my opinion.)
I have suggested to Dr. Chase that the Rutgers Genetics course (as variable in quality as I've heard it is - some report it being better-taught in the summertime, BTW), or some equivalent, be prerequisites or corequisites for the second semester of Experimental Biochemistry - and may suggest this also for the second semester of General Biochemistry - in light of the many people coming to me needing help on what I, as a geneticist, would consider very basic aspects of the plasmid, RNA, sequencing, and PCR labs. While this would take too much wrangling to get through for next year, he is planning on adding a strong recommendation for such a course to the description of Experimental Biochemistry in the course catalog.
I am willing to spend quite a bit of time giving assistance, as you can see from the scheduling comments on my prior webpages and in my Intro Sheet. Some (as expressed in one anonymously-made comment) may be concerned about whether or not the people I work with are doing enough on their own:
My background: My primary background is in biology, specifically molecular genetics. I am mainly qualified for this lab due to:
Getting in touch: The best means of getting in touch with me is to come by Lipman Hall room 118/119, then try Lipman Hall 202 (the SGI computer lab). (If the building is locked up, try the phone number given below - use the 119 number first in that case.) The second best is to email me (see below for the address), since I check my email several times most days. (Note the points on my tutorials page about not sending me email that's something other than plain text - no attachments!) The third best is to call me at 932-9255 extension 119 (202 if that doesn't work; 207 if at night and neither 119 nor 202 have worked). (Do not assume that I'll receive voicemail; only use this method if I (or someone else) answer(s) the call.) Please note that I really prefer not getting phone calls; contacting me via email or in person is much better. The fourth best is to put a note in my box; it is on the first floor of Lipman Hall.
Office Hours: My sleep cycle is too erratic for posting office hours to work. I will try to let you know what times I will (barring unexpected events) be available (usually I will be in and available a lot immediately prior to when any more-involved lab report is due, especially if I am grading it). The best thing to do is to simply ask whether I will be available at a given time (in other words, make an appointment, preferably via email and also usually preferably in the afternoon or evening; if a lot of people are needing me on a particular day, I may announce to everyone (via my webpage or in lab, usually) that I'll be available then. People who've had me (as a TA) before can tell you that I am willing (unless other obligations, including my own research, academics, and health (e.g., my need for sleep), conflict) to work with people at quite odd times and/or for very long hours (I've spent 20+ hours here helping people on a few days...). (I am actually only required to work 15 hours a week on the lab course (on the average), including both time spent in the lab and time spent outside of the lab doing things like grading reports and exams, proctoring exams, etcetera. Time that I spend outside of said 15 hours, which is the case for most time that I spend helping people, is quite purely voluntary on my part; in other words, if you want/need lots of help, do not piss me off. This is not meant to scare you off from asking for help when you need it - it is meant to discourage you from doing things (e.g., cheating or otherwise hurting your fellow students) that will anger me.) You may need to have a cell phone or something with you to call for me (or, preferably, a friend who is already here!) to open the door for you, if it's after 6 or 7 PM or on a weekend - do not yank on the door to try to get it open! Lab reports will be treated as turned in on the previous weekday if they are in prior to the first of:
Subjective Grade: How I do the subjective grade is to note down when you do something good, and when you do something bad. I will fix a particular starting grade, and doing something bad will decrease your subjective grade below this; doing something good will increase it above this. The starting grade and amount up/down will be determined by whatever gives a final mean or median of 85 and a high of 100. Examples of good and bad things:
I normally find I have more +'s than -'s by the end of a semester. This means that those who do get significant minuses (e.g., a lab group a bit back that left lots of gunk in the pig kidney centrifuge bottles...) will get a rather low subjective grade, and that those who simply don't do much either positive or negative won't get a particularly good one.
Nametags: I have problems remembering people's names (including close friends and relatives, BTW!), especially in a class of 20+ people. This sometimes causes problems with assigning subjective grades. I therefore have everyone fill out and wear a nametag. These should be left in the lab, to avoid losing them. At least for the fall semester, if you forget to wear it, that will (if repeated) be subjective points off; if you lose yours, that will definitely be more subjective points off especially since I bought them with my own money!
Curving: I normally will try to curve to a mean of 85 and a high of 100 (although I normally do not curve down). On lab reports and quizzes, I'll circle the final grade that you'll get for each of them. Such curving does not include extra credit points or points taken off for lateness. I frequently wind up having averages of over 85 (due to not curving down and/or after extra credit points) on lab reports I grade, however, although there do tend to be a few people each year who do extremely badly - generally, those who forget to do/include an entire section of the lab report are going to get a much lower grade than the average.
Page written by Allen Smith (send mail to meatcan2@beatrice.rutgers.edu, substituting easmith for meatcan2 - do not just use the mailto link! - see my antispam page(s) for why). (Amplification: Do not send email to the meatcan2 address! Type easmith instead of meatcan2!)
I am not responsible for any pages linked from these, except for those that I have written. Neither is the Structural Biology Computational Laboratory, the Department of Biochemistry and Microbiology, Cook College, or Rutgers University responsible for (or have any copyright on) pages that I have written. My webpages are not official Rutgers webpages.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
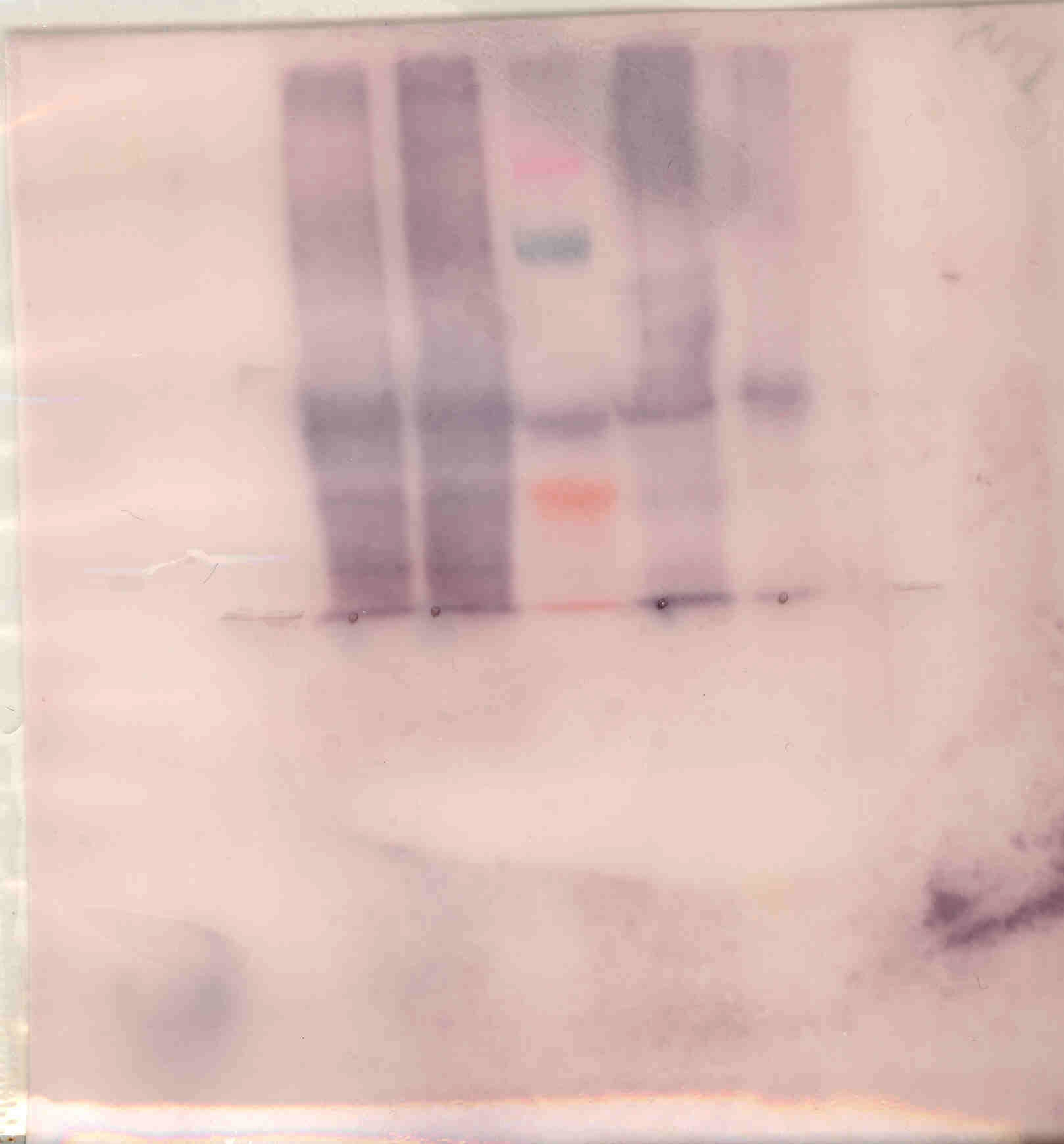{kind=link}
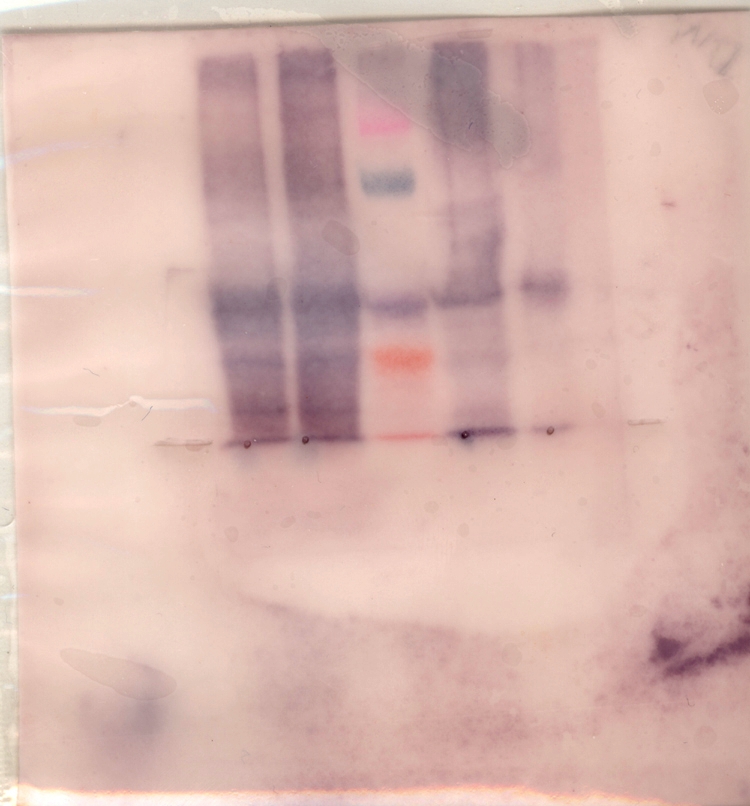{kind=link}
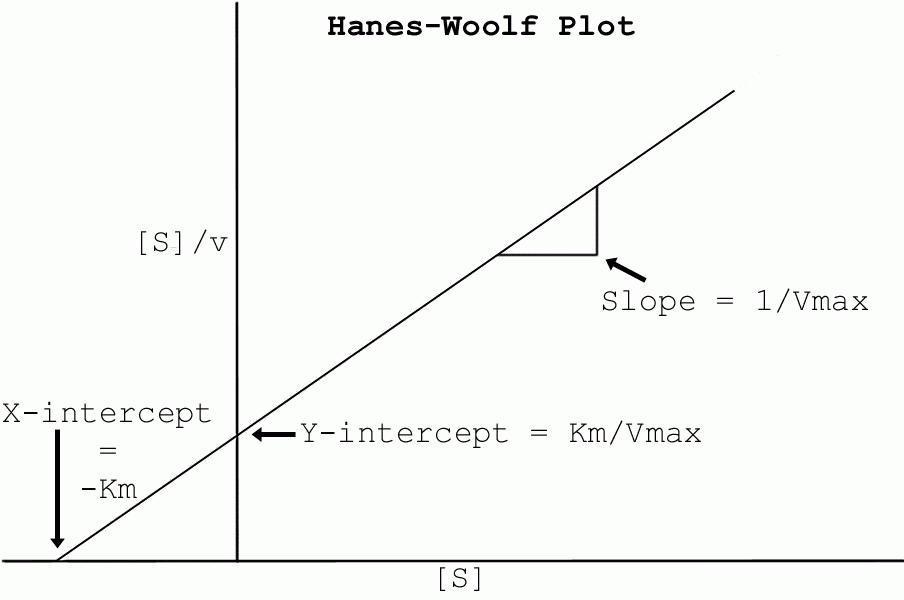{kind=link}